应用数学与交叉科学研究中心生物信息学团队于2024年3月第2次组会按期举行,小组全体成员和各位导师共同参加。在这次组会上,由一名研一员工和两名研二员工分别汇报自己的研究进展,然后老师与同学们对汇报内容进行学术探讨,并对存在的问题给出相应的指导和建议。
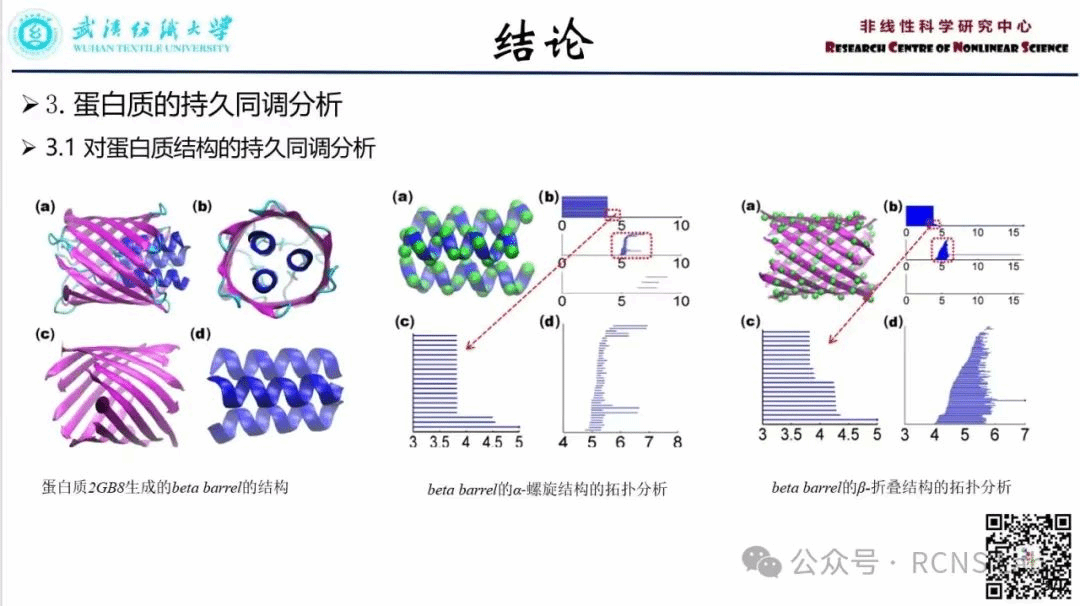
张春焕:本次组会汇报了一篇文献《Persistent homology analysis of protein structure,flexibility and folding》。蛋白质是生物体中最重要的生物分子。了解蛋白质的结构、功能、动力学和转运是生物科学中最具挑战性的任务之一。本文首次将持久司调引入到分子拓扑指纹绞的提取中。分子拓扑指纹被用于蛋白质的表征、鉴定和分类。基于蛋白质紧密度、刚性和连通性之间的相关性,提出了一个由持久拓扑不变量生成的累积条长度,用于蛋白质灵活性的定量建模。并提出了一种基于相关矩阵的过滤方法。这种方法可以准确预测蛋白质b因子分析中使用的最佳特征距离。最后,利用拓扑指纹表征蛋白质折叠过程中的拓扑进化,定量预测蛋白质折叠的稳定性。这项工作揭示了蛋白质的拓扑-功能关系。
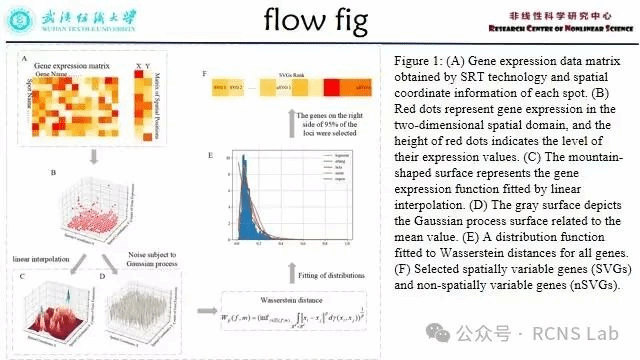
孙睿:本次组会汇报了一篇文献《A spatial variable gene filtrate method based on Wasserstein distance》。识别空间可变基因(SVGs)是连接分子细胞功能与组织表型的关键。空间解析转录组学(SRT)在二维或三维空间坐标上捕获细胞水平的基因表达,可以高效地获得细胞的基因表达数据和空间位置信息。根据SRT获得的数据可以用来识别空间可变基因,然而,目前的计算方法可能无法准确识别关键的SVGs,并且往往无法处理三维空间转录组数据。在这里我们提出了一种识别SVGs的方法(SWDG),通过比较基因表达量分布和其均值分布的wasserstein距离来确定SVGs。该方法的准确性、鲁棒性分别在4个2D数据集和4个3D数据集上得到了验证,其中包含4个真实的实验数据和4个计算机生成的模拟数据。真实的数据集由4种不同的测序方法得到,涉及到生物体的多个组织。
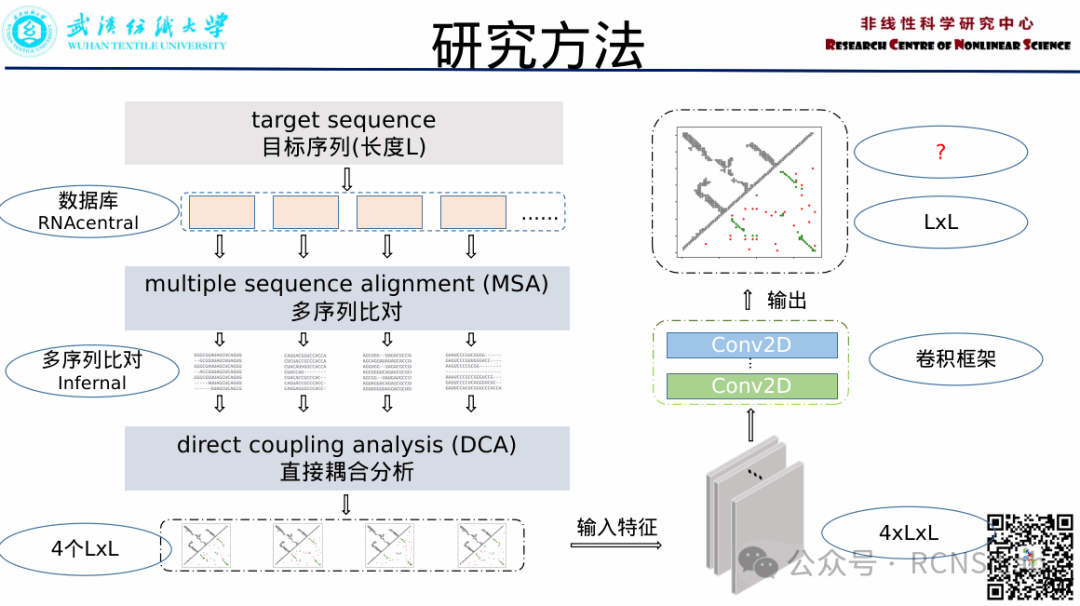
郭成:本次汇报了RNA残基接触预测的近期工作,首先,核糖核酸(RNA)是生物分子的关键分子之一,在细胞中并在许多生物活动中起着重要作用。因为RNA的功能与其三维(3D)结构密切相关,计算方法可以成为一种强大的工具来预测和分析来补充实验工作RNA结构。而间接预测的信息可以集成到计算建模工具中。通过统计方法从RNA家族的多序列比对(MSA)推断出的核苷酸的空间接近信息也可以被利用, 作为空间约束的分子建模工具。在这里我们将核酸序列数据库随机拆分成若干份,分别重新构建序列数据库,分别搜索相似同源家族序列。得到4个多序列比对,通过DCA方法得到4个残基接触结果,最后通过全卷积网络训练来得到一个新的残基接触结果,在训练集和一个测试集上进行测试,结果还存在一定的问题,可能存在的问题还需要再去修改。
— 员工汇报照片展示 —



